完全に国内市場に閉じたビジネスを行っている小規模な医療機器メーカーであればISO13485の対応だけ行ていれば良いかも知れませんが、殆どのグローバルにビジネス展開している医療機器メーカーにとって2~4年おきのFDA監査対応は必須と言えます。このFDA監査で指摘を受け、Warning Letter(警告書)が出され、その対応を間違えば米国に輸出できなくなるだけでなく、多額(28億円)の制裁金を課された日本企業の例すらあるのです。
FDAとは
FDAはCFR(Code of Federal Regulations(連邦行政規則集))の21分冊「Food and Drug」を受け持つ政府機関です。
FDAとは、アメリカ食品医薬品局(Food and Drug Administration)の略称で、 食品などを取り締まるアメリカ合衆国の政府機関。日本の厚生労働省に似た役割
21CFRには1500近い章立てがあり、医療機器メーカーが大きくかかわる規制として820章(QSR:品質システム規制)があります。その他にITシステムの実装としては電子認証(Part11)も関わってくると考えるべきでしょう。
これらのFDA対応は米国市場で医療機器を販売する医療機器メーカーにとって必須であり、日本の基準の基準であるISO13485を取得しているからと言って免れませんし、「規則だからしかたなく対応する」といった考え方では通用しません。
品質を戦略と捉え、「CAPAプロセスなどを通して品質を改善していく」アプローチが求められる。
ITの導入で要求されるFDA対応とは
それでは、ITの側面で実際のFDA査察に備えて考慮しておかなければならない項目を確認しておきたいと思います。
QSRへの対応
QSRが品質システムとなっているため、品質保証部などの一部の問題である、と捉えられがちですが実際には設計、原材料の仕入れから製造、流通、販売と医療機器メーカーの殆どの業務プロセスを通した品質管理が求められます。
「ERPの導入だから品質管理は関係ない」とは言えず、実際にFDAの査察官向けのQSIT(査察ガイド)では以下の領域を重点的に見ることになっています。
- 設計管理
- マネージメントコントロール
- CAPA(Corrective Action; Preventive Action)
- 生産及び工程管理(P&PC)
従って、医療機器メーカーがERPを導入しようとする場合、
通常の調達・生産・在庫・物流・販売などのロジスティクス関連のモジュールと設計監理や工程管理、CAPAなどを切り離して考えることは出来ない。むしろ、設計(DHF)と生産(DHR)、及びDMR(機器原簿)が紐づく形で記録を残す必要がある。
Part11対応
現代のITシステムではログイン時の様々な認証方式が採用されており、「技術的に個人を特定する」という観点では問題なさそうです。しかし、日本のERESガイドラインやアメリカの 21 CFR Part11 で認められている認証方式には制限があります。
また、電子署名法に起源がある日本のERESガイドラインと米国の Part11 とでも違いがありますので、単にパッケージベンダーの決めた認証方式を鵜呑みにも出来ない事情があります。
CAPA対応
CAPA(Corrective Action; Preventive Action)は日本語で是正予防措置とされるため、誤解されやすいと思うのですが、特にCA(是正措置)は品質基準を満たしていない(半)製品の手直しをすることではなく、「2度と同じ場所(工程)で同じ問題が起こらないようにすること」です。
従って、問題の根本原因を追及し、その根本原因に根本的な対処をする必要がありますので、多くの場合設計に行きつくのです。そして、その根本原因が引き起こす問題を内在する設計を見直そうとすると、多くの場合類似・他製品の設計にも影響する可能性が高く、そこまでやってCAPAのプロセスを実施していると言えます。
CSV対応
たとえビジネス系のシステムであるERPなどであっても、上記の様に品質(FDA監査)に密接に関連していますので、当然バリデーションの対象となります。そして、ここは同様の医療機器メーカーや医薬品メーカーでの経験・実績があるITベンダーを選定する必要があります。(経験がものをいう部分です)
また、昨今はERPでもAWSやAzure、GCPといったクラウド環境で使うことが増えてきていますが、このようなクラウド環境(IaaS/PaaSはクラウドベンダーが対応していると聞いていますが、特にSaaSで使用する場合)でCSV対応が可能か具体的の確認しておくことを強くお勧めします。
上記以外
上記は医療機器メーカーへのIT導入において特に注意を要する点ですが、当然これらだけで十分ということではありません。むしろ、上記を踏まえた人の命に関わる医療機器の品質に関する考え方全般を十分理解し、ITシステムとして実装する必要あがります。

RFPとは
情報システムの導入や業務委託を行うにあたり、発注先候補の事業者に具体的な提案を依頼する文書。 システムの目的や概要、要件や制約条件などが記述されている。
とIT用語辞典ではなっていますが、要はITベンダーに対して提案を依頼する文書です。この時、自社が何をやりたいのか(どの様なシステムを導入したいのか)、提案にあたり満たして欲しい要件など、自社の情報を前提としてある程度提示した上で提案を受けないと意図した内容と全く違ったり、ベンダー間の比較が出来なくなってしまいます。
従って、RFPの作成はそれ自体が自社のIT基本構想を作成して纏めたものであり、この時点である程度の基本構想・業務設計を行うと考えたほうが良いかと思います。
医療機器メーカーがRFP作成を依頼する先が無い理由
RFP作成は中立の立場が基本
医療機器メーカーがRFP作成を依頼する先として可能性があるとすれば下記の様な相手かと思います。
- FDA監査・QSR専門コンサルティング会社
- IT戦略コンサルティング
- ITベンダー
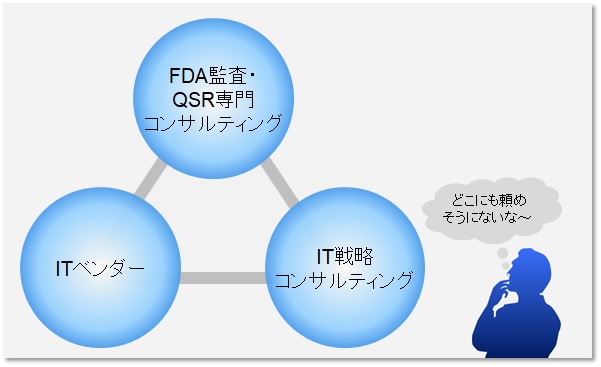
◆ FDA監査・QSR専門コンサルティング会社
これらの企業は、ほぼアメリカに本社を置きFDAの規制を熟知(元FDA監査員だったりしますので当然です)している会社で、どの様に対応すればFDA監査に無難に対応出来るかコンサルティングする会社です。
どの様な規制業界にもありがちですが、実際に米国で医療(機器・薬)ビジネスを行うには頼らざるを得ない存在です。しかし、ほぼアメリカから出張ベースで全世界を旅しているような人達ですので、彼らが個別企業のITツール選定のためのRFP作成やITベンダー選定のコンサルティングを現実的な価格でサービスしてくれるとはとても思えません。
実際のITツールにも詳しくはないと思われます。(FDA対応の為にどうすればクリアし易いかのアドバイスはしてくれるかもしれません)
◆ IT戦略コンサルティング
IT戦略ファームと言われる、IT導入を行わないコンサルティング会社が多く存在します。これらのコンサルティング会社は中立な立場でIT戦略を策定はしてくれます。しかし、個別(医療機器)業界の特殊なFDA規制などに詳しいことはまずないと考えたほうが良いかと思います。
また、相当な金額となりますので、ITベンダー選定と言うよりは、企業としてのIT全体の戦略をお願いするのが妥当かと思います。
◆ ITベンダー
これらの内、最もRFP作成を依頼してはいけない先は当然ITベンダーです。これから提案を依頼し、選定しようとしている相手にRFPの作成自体を依頼してはいけません。殆ど無料でRFP作成を請け負うと言ってくるかも知れませんが、「ただより高いものはない」と考えるべきです。
確実に紐付きになり、そのIT会社に都合が良いRFPを作成され、後々高額を搾取されるはめになるはずです。

結局RFP策定を依頼出来る会社が無い
結局は適切なRFP作成を依頼できる適切な依頼先が無いのが現実で、それだけ特殊な業界だとも言えます。やはり、完全に独立性を保った中立な立場でRFP作成やITベンダー選定を出来る会社を探す必要があります。











